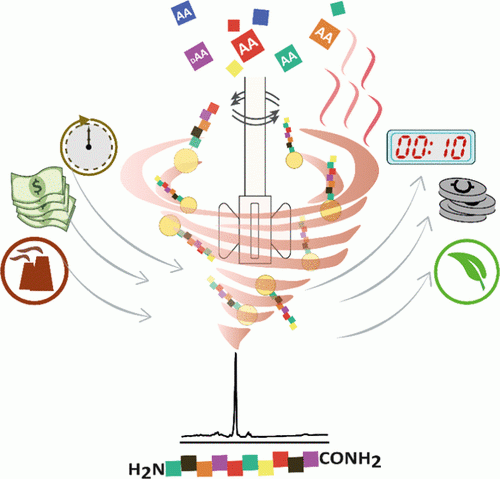
Main observation and conclusion A green and efficient photolabile protecting group (PPG)-mediated glycosidation approach for the synthesis of 2-deoxy-glycosides is reported. By employing ortho-nitrobenzyl carbonate (oNBC) as PPG, N,N-dimethylformamide (DMF)-modulated SPhosAuNTf2-promoted glycosidation with per-oNBC-protected 2-deoxy-glycosyl ynenoates afforded the 2-deoxy-glycosides with moderate to excellent α-selectivities presumably depending on the reactivities of the acceptor alcohols. Based on the PPG-mediated glycosidation approach, oligosaccharides with three to six oNBC groups were effectively achieved. The multiple oNBC groups in the 2-deoxy-glycosides were completely cleaved by irradiation at 365 nm, resulting in the desired 2-deoxy-glycosides in an efficient manner without affecting the common alkyne, alkene and azide functional groups and the traditional protecting groups on the aglycones. This article is protected by copyright. All rights reserved.Main observation and conclusion A green and efficient photolabile protecting group (PPG)-mediated glycosidation approach for the synthesis of 2-deoxy-glycosides is reported. By employing ortho-nitrobenzyl carbonate (oNBC) as PPG, N,N-dimethylformamide (DMF)-modulated SPhosAuNTf2-promoted glycosidation with per-oNBC-protected 2-deoxy-glycosyl ynenoates afforded the 2-deoxy-glycosides with moderate to excellent α-selectivities presumably depending on the reactivities of the acceptor alcohols. Based on the PPG-mediated glycosidation approach, oligosaccharides with three to six oNBC groups were effectively achieved. The multiple oNBC groups in the 2-deoxy-glycosides were completely cleaved by irradiation at 365 nm, resulting in the desired 2-deoxy-glycosides in an efficient manner without affecting the common alkyne, alkene and azide functional groups and the traditional protecting groups on the aglycones. This article is protected by copyright. All rights reserved.

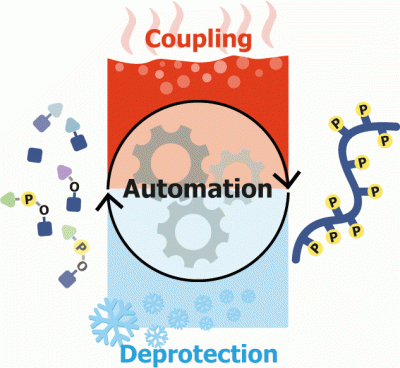
Developing automated platforms is essential for accelerating the preparation of bioactive compound libraries. After a decade of biopolymer synthesis, it is clear that automating these processes was pivotal to the development of biochemistry and chemical biology for advancing medicinal chemistry and for providing new diagnoses and therapeutic tools. Synthesis of glycans and glycoconjugates is far more complicated than the other biopolymer families. Automating glycan synthesis has been a long and hard journey which is still ongoing. This chapter focuses on the development of a commercial platform, Glyconeer™, aimed to automate the synthesis of glycans. The design and process leading to the establishment of the current setup are described. The unique considerations required from a system that is suitable for glycan synthesis are the focus of the chapter. We explain how the selected setup architecture and its unique features comply with the unusual demands of automated solid phase synthesis of glycans. We will present the common modules, building blocks, and chemistries used by the synthesizer. Preparation, handling, and running of both software and hardware are presented from the user point of view. A critical view of the limitations and advantages of the system is aimed to provide a roadmap for future improvement.
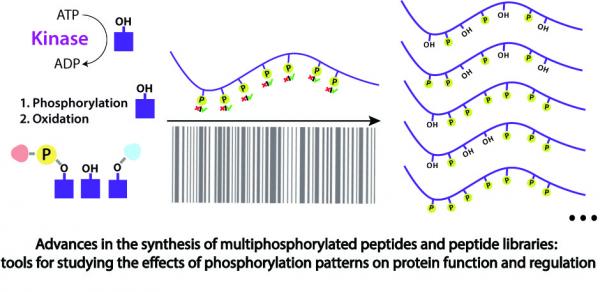
Abstract Multi phosphorylated peptides are key tools in understanding the biological roles of protein phosphorylation patterns. In this work, we focused on multi phosphorylated peptides with over four, clustered, phosphorylation sites that are termed herein heavily phosphorylated peptides (HPPs). The synthesis of heavily phosphorylated peptides is extremely difficult and requires the use of a wide temperature range. Standard peptide synthesizers are incapable of both cooling and heating, which impedes the automated synthesis of those peptides. Herein, we used the oligosaccharide synthesizer Glyconeer 2.1 to develop a protocol for the automated synthesis of heavily phosphorylated peptides. The Glyconeer 2.1 is able to both cool and heat, which enabled the development of highly controlled coupling and deprotection conditions that were used for the automated synthesis of four different heavily phosphorylated peptides with five or more, clustered, phosphorylation sites. Our approach paves the way for an easy automated synthesis of a variety of heavily phosphorylated peptides.
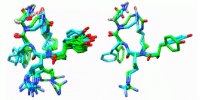
Oxytocin is a neuropeptide that binds copper ions in nature. The structure of oxytocin in interaction with Cu2+ was determined here by NMR, showing which atoms of the peptide are involved in binding. Paramagnetic relaxation enhancement NMR analyses indicated a binding mechanism where the amino terminus was required for binding and subsequently Tyr2, Ile3 and Gln4 bound in that order. The aromatic ring of Tyr2 formed a π-cation interaction with Cu2+. Oxytocin copper complex structure revealed by paramagnetic relaxation enhancement NMR analyses.

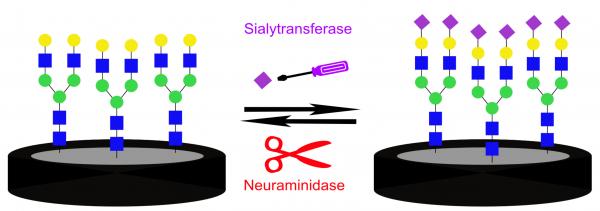
Sialylated glycans and glycoproteins are involved in cellular communication and are crucial for distinguishing between signal pathways. Sialylation levels and patterns modulate recognition events and are regulated by the enzymatic activity of sialyltransferases and neuraminidases. Abnormal activity of these enzymes is related to diseases such as cancer and viral infection. Monitoring these enzymatic activities offers valuable diagnostic tools. This work presents an impedimetric biosensing platform for following and detecting sialylation and desialylation processes. This platform is based on a native biantennary N-glycan substrate attached to a glassy carbon electrode. Changes in the molecular layer, as a result of enzymatic reactions, were detected by electrochemical impedance spectroscopy, displaying high sensitivity to the enzymatic surface reactions. Increase in the molecular layer roughness in response to the sialylation was visualized using atomic force microscopy. After enzymatic sialylation, the presence of sialic acid was confirmed using cyclic voltammetry by coupling of the redox active marker aminoferrocene. The sialylation showed selectivity toward the N-glycan compared to another glycan substrate. A time dependent sialylation was followed by electrochemical impedance spectroscopy, proving that the new system can be applied to evaluate the enzymatic kinetics. Our findings suggest that analyzing sialylation processes using this platform can become a useful tool for the detection of pathological states and pathogen invasion.
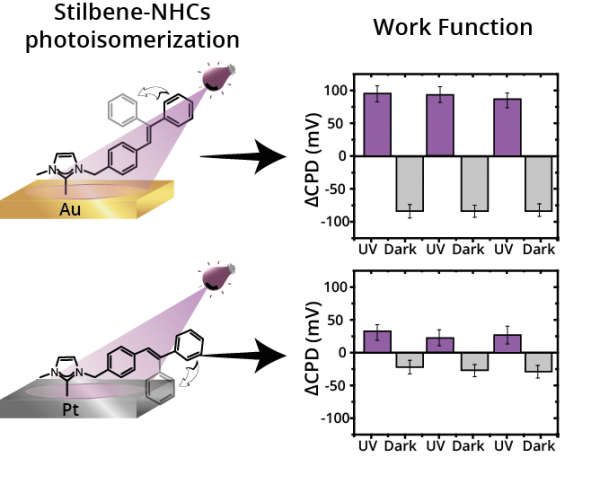
Self-assembly of photo-responsive molecules is a robust technology for reversibly tuning the properties of functional materials. Herein, we probed the crucial role of surface–adsorbate interactions on the adsorption geometry of stilbene-functionalized N-heterocyclic carbenes (stilbene-NHCs) monolayers and its impact on surface potential. Stilbene-NHCs on Au film accumulated in a vertical orientation that enabled high photoisomerization efficiency and reversible changes in surface potential. Strong metal–adsorbate interactions led to flat-lying adsorption geometry of stilbene-NHCs on Pt film, which quenched the photo-isomerization influence on surface potential. It is identified that photo-induced response can be optimized by positioning the photo-active group in proximity to weakly-interacting surfaces.
J. Attia, S. Nir, E. Mervinetsky, D. Balogh, A. Gitlin-Domagalska, I. Alshanski, M. Reches, M. Hurevich, and S. Yitzchaik. 2021. “
Non-covalently embedded oxytocin in alkanethiol monolayer as Zn2+ selective biosensor.” Scientific Reports, 11, 1.
Abstract Peptides are commonly used as biosensors for analytes such as metal ions as they have natural binding preferences. In our previous peptide-based impedimetric metal ion biosensors, a monolayer of the peptide was anchored covalently to the electrode. Binding of metal ions resulted in a conformational change of the oxytocin peptide in the monolayer, which was measured using electrochemical impedance spectroscopy. Here, we demonstrate that sensing can be achieved also when the oxytocin is non-covalently integrated into an alkanethiol host monolayer. We show that ion-binding cause morphological changes to the dense host layer, which translates into enhanced impedimetric signals compared to direct covalent assembly strategies. This biosensor proved selective and sensitive for Zn2+ ions in the range of nano- to micro-molar concentrations. This strategy offers an approach to utilize peptide flexibility in monitoring their response to the environment while embedded in a hydrophobic monolayer.

 mediated interleukin-8 glycan binding")






 complex using paramagnetic relaxation enhancement NMR analysis")











 Ions with GGH Peptide Realized with Si-Nanoribbon ISFET")