Maor, Inbar, Naama Koifman, Ellina Kesselman, Pnina Matsanov, Ilan Shumilin, Daniel Harries, and Iris Sonia Weitz. “
Molecular Self-Assembly under Nanoconfinement: Indigo Carmine Scroll Structures Entrapped Within Polymeric Capsules.”
Nanoscale 13 (2021): 20462-20470.
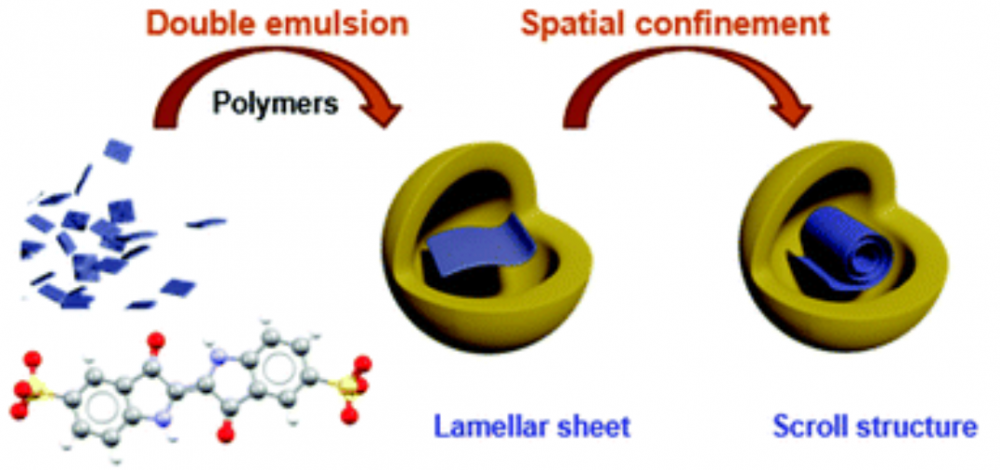
Publisher's VersionAbstractMolecular self-assembly forms structures of well-defined organization that allow control over material properties, affording many advanced technological applications. Although the self-assembly of molecules is seemingly spontaneous, the structure into which they assemble can be altered by carefully modulating the driving forces. Here we study the self-assembly within the constraints of nanoconfined closed spherical volumes of polymeric nanocapsules, whereby a mixture of polyester-polyether block copolymer and methacrylic acid methyl methacrylate copolymer forms the entrapping capsule shell of nanometric dimensions. We follow the organization of the organic dye indigo carmine that serves as a model building unit due to its tendency to self-assemble into flat lamellar molecular sheets. Analysis of the structures formed inside the nanoconfined space using cryogenic-transmission electron microscopy (cryo-TEM) and cryogenic-electron tomography (cryo-ET) reveal that confinement drives the self-assembly to produce tubular scroll-like structures of the dye. Combined continuum theory and molecular modeling allow us to estimate the material properties of the confined nanosheets, including their elasticity and brittleness. Finally, we comment on the formation mechanism and forces that govern self-assembly under nanoconfinement.
Allolio, Christoph, and Daniel Harries. “
Calcium Ions Promote Membrane Fusion by Forming Negative-Curvature Inducing Clusters on Specific Anionic Lipids.”
ACS Nano 15, no. 8 (2021): 12880–12887.
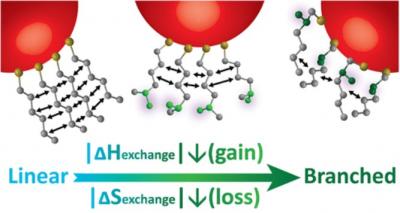
Publisher's VersionAbstractVesicles enriched in certain negatively charged lipids, such as phosphatidylserine and PIP2, are known to undergo fusion in the presence of calcium ions without assistance from protein assemblies. Other lipids do not exhibit this propensity, even if they are negatively charged. Using our recently developed methodology, we extract elastic properties of a representative set of lipids. This allows us to trace the origin of lipid-calcium selectivity in membrane fusion to the formation of lipid clusters with long-range correlations that induce negative curvature on the membrane surface. Furthermore, the clusters generate lateral tension in the headgroup region at the membrane surface, concomitantly also stabilizing negative Gaussian curvature. Finally, calcium binding also reduces the orientational polarization of water around the membrane head groups, potentially reducing the hydration force acting between membranes. Binding calcium only weakly increases membrane bending rigidity and tilt moduli, in agreement with experiments. We show how the combined effects of calcium binding to membranes lower the barriers along the fusion pathway that lead to the formation of the fusion stalk as well as the fusion pore.

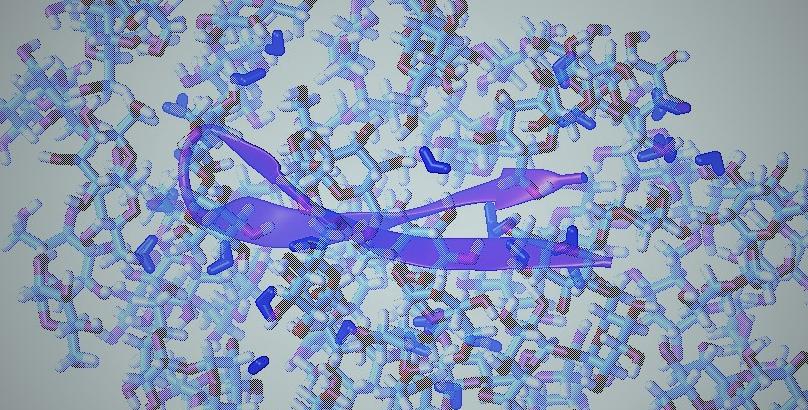
Olgenblum, Gil I., Liel Sapir, Frank Wien, and Daniel Harries. “
β-Hairpin Miniprotein Stabilization in Trehalose Glass Is Facilitated by an Emergent Compact Non-Native State.”
The Journal of Physical Chemistry Letters 12, no. 32 (2021): 7659–7664.
Publisher's VersionAbstractFrom stem cell freeze-drying to organ storage, considerable recent efforts have been directed toward the development of new preservation technologies. A prominent protein stabilizing strategy involves vitrification in glassy matrices, most notably those formed of sugars such as the biologically relevant preservative trehalose. Here, we compare the folding thermodynamics of a model miniprotein in solution and in the glassy state of the sugars trehalose and glucose. Using synchrotron radiation circular dichroism (SRCD), we find that the same native structure persists in solution and glass. However, upon transition to the glass, a completely different, conformationally restricted unfolded state replaces the disordered denatured state found in solution, potentially inhibiting misfolding. Concomitantly, a large exothermic contribution is observed in glass, exposing the stabilizing effect of interactions with the sugar matrix on the native state. Our results shed light on the mechanism of protein stabilization in sugar glass and should aid in future preservation technologies.

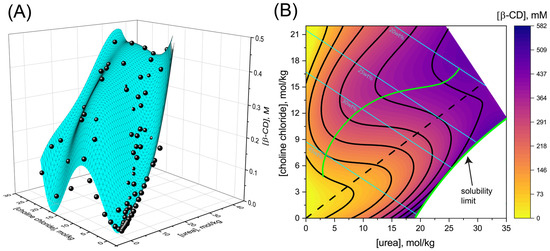
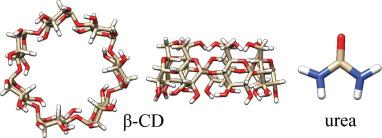
Shumilin, Ilan, and Daniel Harries. “
Cyclodextrin solubilization in hydrated reline: Resolving the unique stabilization mechanism in a deep eutectic solvent.”
The Journal of Chemical Physics 154, no. 22 (2021): 224505.
Publisher's VersionAbstractBy complexing with hydrophobic compounds, cyclodextrins afford increased solubility and thermodynamic stability to hardly soluble compounds, thereby underlining their invaluable applications in pharmaceutical and other industries. However, common cyclodextrins such as β-cyclodextrin, suffer from limited solubility in water, which often leads to precipitation and formation of unfavorable aggregates, driving the search for better solvents. Here, we study the solvation of cyclodextrin in deep eutectic solvents (DESs), environmentally friendly media that possess unique properties. We focus on reline, the DES formed from choline chloride and urea, and resolve the mechanism through which its constituents elevate β-cyclodextrin solubility in hydrated solutions compared to pure water or dry reline. Combining experiments and simulations, we determine that the remarkable solubilization of β-cyclodextrin in hydrated reline is mostly due to the inclusion of urea inside β-cyclodextrin’s cavity and at its exterior surfaces. The role of choline chloride in further increasing solvation is twofold. First, it increases urea’s solubility beyond the saturation limit in water, ultimately leading to much higher β-cyclodextrin solubility in hydrated reline in comparison to aqueous urea solutions. Second, choline chloride increases urea’s accumulation in β-cyclodextrin’s vicinity. Specifically, we find that the accumulation of urea becomes stronger at high reline concentrations, as the solution transitions from reline-in-water to water-in-reline, where water alone cannot be regarded as the solvent. Simulations further suggest that in dry DES, the mechanism of β-cyclodextrin solvation changes so that reline acts as a quasi-single component solvent that lacks preference for the accumulation of urea or choline chloride around β-cyclodextrin.
By complexing with hydrophobic compounds, cyclodextrins afford increased solubility and thermodynamic stability to hardly soluble compounds, thereby underlining their invaluable applications in pharmaceutical and other industries. However, common cyclodextrins such as β-cyclodextrin, suffer from limited solubility in water, which often leads to precipitation and formation of unfavorable aggregates, driving the search for better solvents. Here, we study the solvation of cyclodextrin in deep eutectic solvents (DESs), environmentally friendly media that possess unique properties. We focus on reline, the DES formed from choline chloride and urea, and resolve the mechanism through which its constituents elevate β-cyclodextrin solubility in hydrated solutions compared to pure water or dry reline. Combining experiments and simulations, we determine that the remarkable solubilization of β-cyclodextrin in hydrated reline is mostly due to the inclusion of urea inside β-cyclodextrin’s cavity and at its exterior surfaces. The role of choline chloride in further increasing solvation is twofold. First, it increases urea’s solubility beyond the saturation limit in water, ultimately leading to much higher β-cyclodextrin solubility in hydrated reline in comparison to aqueous urea solutions. Second, choline chloride increases urea’s accumulation in β-cyclodextrin’s vicinity. Specifically, we find that the accumulation of urea becomes stronger at high reline concentrations, as the solution transitions from reline-in-water to water-in-reline, where water alone cannot be regarded as the solvent. Simulations further suggest that in dry DES, the mechanism of β-cyclodextrin solvation changes so that reline acts as a quasi-single component solvent that lacks preference for the accumulation of urea or choline chloride around β-cyclodextrin.

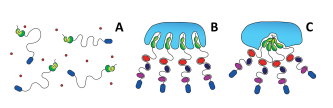
Garfagnini, Tommaso, Yael Levi-Kalisman, Daniel Harries, and Assaf Friedler. “
Osmolytes and crowders regulate aggregation of the cancer-related L106R mutant of the Axin protein.”
Biophysical J. 120, no. 16 (2021): 3455-3469.
Publisher's VersionAbstractProtein aggregation is involved in a variety of diseases, including neurodegenerative diseases and cancer. The cellular environment is crowded by a plethora of cosolutes comprising small molecules and biomacromolecules at high concentrations, which may influence the aggregation of proteins in vivo. To account for the effect of cosolutes on cancer-related protein aggregation, we studied their effect on the aggregation of the cancer-related L106R mutant of the Axin protein. Axin is a key player in the Wnt signaling pathway, and the L106R mutation in its RGS domain results in a native molten globule that tends to form native-like aggregates. This results in uncontrolled activation of the Wnt signaling pathway, leading to cancer. We monitored the aggregation process of Axin RGS L106R in vitro in the presence of a wide ensemble of cosolutes including polyols, amino acids, betaine and polyethylene glycol (PEG) crowders. Except myo-inositol, all polyols decreased RGS L106R aggregation, with carbohydrates exerting the strongest inhibition. Conversely, betaine and PEGs enhanced aggregation. These results are consistent with the reported effects of osmolytes and crowders on the stability of molten globular proteins and with both amorphous and amyloid aggregation mechanisms. We suggest a model of Axin L106R aggregation in vivo, whereby molecularly small osmolytes keep the protein as a free solublemolecule but the increased crowding of the bound state by macromolecules induces its aggregation at the nano-scale. To our knowledge, this is the first systematic study on the effect of osmolytes and crowders on a process of native-like aggregation involved in pathology, as it sheds light on the contribution of cosolutes to the onset of cancer as a protein misfolding disease, and on the relevance of aggregation in the molecular aetiology of cancer.