Fink, Lea, Christoph Allolio, Jehuda Feitelson, Carmen Tamburu, Daniel Harries, and Uri Raviv. “
Bridges of Calcium Bicarbonate Tightly Couple Dipolar Lipid Membranes.”
Langmuir 36, no. 36 (2020): 10715–10724.
Publisher's VersionAbstractThe interaction between lipid membranes and ions is associated with a range of key physiological processes. Most earlier studies have focused on the interaction of lipids with cations, while the specific effects of the anions have been largely overlooked. Owing to dissolved atmospheric carbon dioxide, bicarbonate is an important ubiquitous anion in aqueous media. In this paper, we examined the effect of bicarbonate anions on the interactions between dipolar lipid membranes in the presence of previously adsorbed calcium cations. Using a combination of solution X-ray scattering, osmotic stress, and molecular dynamic simulations, we followed the interactions between 1,2-didodecanoyl-sn-glycero-3-phosphocholine (DLPC) lipid membranes that were dialyzed against CaCl2 solutions in the presence and absence of bicarbonate anions. Calcium cations adsorbed onto DLPC membranes charge them and lead to their swelling. In the presence of bicarbonate anions, however, the calcium cations can tightly couple one dipolar DLPC membrane to the other and form a highly condensed and dehydrated lamellar phase with a repeat distance of 3.45±0.02 nm. Similar tight condensation and dehydration has only been observed between charged membranes in the presence of multivalent counterions. Bridging between bilayers by calcium bicarbonate complexes induced this arrangement. Furthermore, in this condensed phase the lipid molecules and the adsorbed ions were arranged in a 2D oblique lattice.

Schachter, Itay, Christoph Allolio, George Khelashvili, and Daniel Harries. “
Confinement in Nanodiscs Anisotropically Modifies Lipid Bilayer Elastic Properties.”
Journal of Physical Chemistry B 124, no. 33 (2020): 7166–7175.
Publisher's VersionAbstract
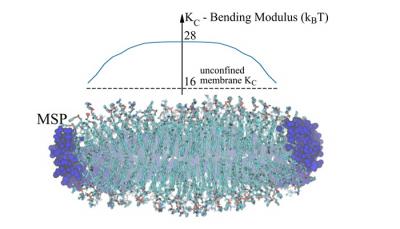
Lipid nanodiscs are small synthetic lipid bilayer structures that are stabilized in solution by special circumscribing (or scaffolding) proteins or polymers. Because they create native-like environments for transmembrane proteins, lipid nanodiscs have become a powerful tool for structural determination of this class of systems when combined with cryo-electron microscopy or nuclear magnetic resonance. The elastic properties of lipid bilayers determine how the lipid environment responds to membrane protein perturbations, and how the lipid in turn modifies the conformational state of the embedded protein. However, despite the abundant use of nanodiscs in determining membrane protein structure, the elastic material properties of even pure lipid nanodiscs (i.e., without embedded proteins) have not yet been quantitatively investigated. A major hurdle is due to the inherently non-local treatment of the elastic properties of lipid systems implemented by most existing methods, both experimental and computational. In addition, these methods are best suited for very large “infinite” size lipidic assemblies, or ones that contain periodicity, in the case of simulations. We have previously described a computational analysis of molecular dynamics simulations designed to overcome these limitations, so that it allows quantification of the bending rigidity (KC) and tilt moduli (κt) on a local scale even for finite, non-periodic systems, such as lipid nanodiscs. Here we use this computational approach to extract values of KC and κt for a set of lipid nanodisc systems that vary in size and lipid composition. We find that the material properties of lipid nanodiscs are different from those of infinite bilayers of corresponding lipid composition, highlighting the effect of nanodisc confinement. Nanodiscs tend to show higher stiffness than their corresponding macroscopic bilayers, and moreover, their material properties vary spatially within them. For small-size MSP1 nanodiscs, the stiffness decreases radially, from a value that is larger in their center than the moduli of the corresponding bilayers by a factor of ~2-3. The larger nanodiscs (MSP1E3D1 and MSP2N2) show milder spatial changes of moduli that are composition dependent and can be maximal in the center or at some distance from it. These trends in moduli correlate with spatially varying structural properties, including the area per lipid and the nanodisc thickness. Finally, as has previously been reported, nanodiscs tend to show deformations from perfectly flat circular geometries to varying degrees, depending on size and lipid composition. The modulations of lipid elastic properties that we find should be carefully considered when making structural and functional inferences concerning embedded proteins.
Shumilin, Ilan, Benny Bogoslavsky, and Daniel Harries. “
Stressing Crystals With Solutes: Effects of Added Solutes on Crystalline Caffeine and their Relevance to Determining Transfer Free Energies.”
Colloids and Surfaces A: Physicochemical and Engineering Aspects 599 (2020): 124889.
Publisher's VersionAbstractIn calculating transfer free energies of solvated substances, the coexisting crystal state is often taken as the reference. Furthermore, the free energy of this reference state is often assumed to remain constant upon changes made in solution. Yet little is known about the way added cosolutes impact the thermodynamic stability of the out-of-solution crystal phase. To provide insight into the changes in the activity of the coexisting solid state, we used caffeine, a well-studied hydrophobic compound that forms a hydrated crystal in saturated aqueous solutions. By using X-ray powder diffraction, we found that cosolutes, such as trehalose, sucrose, sodium sulfate, and polyethylene glycol (PEG) alter the unit cell volume of crystalline caffeine, in a concentration dependent manner. The dehydration of solid caffeine translates into an overall increase in its free energy, which can be directly calculated as the reversible ΠV work required to compress the crystal. We determined that trehalose, sucrose, and sodium sulfate increase the free energy of the solid, while PEG decreases it. For 2 mol/kg trehalose, this change in free energy corresponds to 17 % of the total change in solvation free energy of monomeric caffeine. Although our results indicate that cosolutes modify the free energy of the solid less than that of the solvated state, this effect is non negligible and measurable, suggesting that it should generally be taken into account as a contribution to changes in solubility, particularly whenever the solid phase is hydrated.

Sapir, Liel, and Daniel Harries. “
Restructuring a Deep Eutectic Solvent by Water: The Nanostructure of Hydrated Choline Chloride/Urea.”
Journal of Chemical Theory and Computation 16 (2020): 3335-3342.
Publisher's VersionAbstractDeep eutectic mixtures are a promising sustainable and diverse class of tunable solvents that hold great promise for various green chemical and technological processes. Many deep eutectic solvents (DES) are hygroscopic and find use in applications with varying extents of hydration, hence urging a profound understanding of changes in the nanostructure of DES with water content. Here, we report on molecular dynamics simulations of the quintessential choline chloride–urea mixture, using a newly parametrized force field with scaled charges to account for physical properties of hydrated DES mixtures. These simulations indicate that water changes the nanostructure of solution even at very low hydration. We present a novel approach that uses convex constrained analysis to dissect radial distribution functions into base components representing different modes of local association. Specifically, DES mixtures can be deconvoluted locally into two dominant competing nanostructures, whose relative prevalence (but not their salient structural features) change with added water over a wide concentration range, from dry up to ∼30 wt % hydration. Water is found to be associated strongly with several DES components but remarkably also forms linear bead-on-string clusters with chloride. At high water content (beyond ∼50 wt % of water), the solution changes into an aqueous electrolyte-like mixture. Finally, the structural evolution of the solution at the nanoscale with extent of hydration is echoed in the DES macroscopic material properties. These changes to structure, in turn, should prove important in the way DES acts as a solvent and to its interactions with additive components.

Olgenblum, Gil I., Liel Sapir, and Daniel Harries. “
Properties of Aqueous Trehalose Mixtures: Glass Transition and Hydrogen Bonding.”
Journal of Chemical Theory and Computation 16 (2020): 1249-1262.
Publisher's VersionAbstractTrehalose is a naturally occurring disaccharide known to remarkably stabilize biomacromolecules in the biologically active state. The stabilizing effect is typically observed over a large concentration range and affects many macromolecules including proteins, lipids, and DNA. Of special interest is the transition from aqueous solution to the dense and highly concentrated glassy state of trehalose that has been implicated in bioadaptation of different organisms toward desiccation stress. Although several mechanisms have been suggested to link the structure of the low water content glass with its action as an exceptional stabilizer, studies are ongoing to resolve which are most pertinent. Specifically, the role that hydrogen bonding plays in the formation of the glass is not well resolved. Here we model aqueous trehalose mixtures over a wide concentration range, using molecular dynamics simulations with two available force fields. Both force fields indicate glass transition temperatures and osmotic pressures that are close to experimental values, particularly at high trehalose contents. We develop and employ a methodology that allows us to analyze the thermodynamics of hydrogen bonds in simulations at different water contents and temperatures. Remarkably, this analysis is able to link the liquid to glass transition with changes in hydrogen bond characteristics. Most notably, the onset of the glassy state can be quantitatively related to the transition from weakly to strongly correlated hydrogen bonds. Our findings should help resolve the properties of the glass and the mechanisms of its formation in the presence of added macromolecules.